Point de pratique
La maladie de Kawasaki : des directives cliniques sur le diagnostic et la prise en charge

Affichage : le 2 février 2026
Auteur(s) principal(aux)
Audrea Chen MD, Evelyn Rozenblyum MD, Nagib Dahdah MD, Bianca Lang MD, Rosie Scuccimarri MD, Piya Lahiry M. Sc. Ph. D. MD, Herman Tam MD, Mercedes Chan MD, Andrea Human MD, Jennifer Lee MD M. Sc., Peter Wong MD, Paul Tsoukas MD, Rae SM Yeung MD; Société canadienne de pédiatrie, Comité de la pédiatrie communautaire
Résumé
La maladie de Kawasaki est une vasculite systémique qui doit être traitée rapidement pour éviter des anévrismes coronaires et les complications cardiaques qui en découlent. Le présent point de pratique propose des directives pour diagnostiquer la maladie de Kawasaki et la prendre en charge, y compris dans les scénarios de maladie grave et de risque marqué d’anévrisme coronaire qui justifient des consultations en surspécialité.
Mots-clés : anévrisme coronaire; antiplaquettaire; échocardiographie, IgIV; maladie de Kawasaki
Contexte
La maladie de Kawasaki (MK) est une vasculite systémique des petites et moyennes artères[1][2]. Au Canada, son incidence est plus élevée chez les enfants d’âge préscolaire (de 19,6 à 26,0 cas sur 100 000 enfants de moins de cinq ans), mais la maladie peut se manifester tout au long de l’enfance[3]-[5].
La MK est une cause importante de cardiopathie infantile acquise[1]-[5]. Les lésions coronaires, y compris les dilatations et les anévrismes, en sont les séquelles les plus graves (chez 20 % à 25 % des patients non traités)[6]. Bien qu’elles soient rares (moins de 1 %), les complications des anévrismes coronaires incluent la thrombose, l’ischémie myocardique et la rupture[1][6][7].
Le diagnostic
Les critères cliniques (tableau 1) permettent de confirmer le diagnostic, même si certains ne sont répertoriés qu’au moment d’établir l’histoire médicale de l’enfant. On doit envisager une MK incomplète en présence de fièvre et de moins de quatre des critères principaux ou chez les nourrissons (âgés de moins de 12 mois) atteints d’une fièvre prolongée et inexpliquée[1]. D’autres diagnostics doivent également être envisagés (tableau 2). La MK peut se manifester conjointement avec une infection, auquel cas les deux affections doivent être traitées[8]. Si la MK est accompagnée d’un choc, il faut envisager la possibilité d’un sepsis.
|
Tableau 1. Les caractéristiques cliniques de la maladie de Kawasaki |
|
|
Critères principaux d’une MK complète |
Fièvre ≥5 jours* et au moins 4 des critères suivants :
*En présence de ≥4 principaux critères cliniques, le diagnostic peut être posé au 4e jour de fièvre. |
|
Critères de MK incomplète[1] |
ET une VS ≥40 mm/h OU une CRP ≥30 mg/L Les nourrissons et les enfants qui respectent les critères précédents doivent présenter :
OU 3 des 6 résultats suivants :
|
|
Éruption périnéale desquamative, hydrops de la vésicule biliaire, méningite aseptique, inflammation au foyer d’un vaccin BCG antérieur, uvéite, œdème rétropharyngé, arthrite |
|
ALT alanine aminotransférase; BCG bacille Calmette-Guérin; cm centimètre; CRP protéine C réactive; hpf champ à fort grossissement; L litre; mg milligramme; MK maladie de Kawasaki; mm millimètre; VS vitesse de sédimentation érythrocytaire
Information tirée de la référence 8
L’échocardiographie
L’échocardiographie coronaire est essentielle à la prise en charge de la MK, mais il ne faut pas en attendre les résultats pour poser le diagnostic et entreprendre le traitement[1][2].
L’interprétation du diamètre de l’artère coronaire repose sur les calculs du score Z[9]. Les catégories de lésions coronaires incluent une dilatation (Z ≥2 à <2,5), un petit anévrisme (Z ≥2,5 à <5), un anévrisme moyen (Z ≥5 à <10) ou un anévrisme gros ou géant (Z ≥10, ou diamètre ≥8 mm)[10]. Le pronostic cardiaque s’aggrave proportionnellement à l’augmentation des dimensions des artères coronaires[10]. Il faut procéder à des échocardiographies sérielles pour surveiller les artères coronaires en raison du risque d’anévrismes jusqu’à quatre à six semaines après l’apparition de la maladie[1][2].
Les immunoglobulines intraveineuses
Une dose d’immunoglobulines intraveineuses (IgIV; 2 g/kg) réduit le risque d’anévrisme coronaire à moins de 5 % lorsqu’elle est administrée dans les dix jours suivant l’apparition de la fièvre[1]. En cas de soupçons de MK, les patients devraient tout de même être traités après le dixième jour lorsque la fièvre et l’inflammation persistent[1]. La fièvre devrait s’être résorbée de 24 à 36 heures après la fin du traitement aux IgIV. Les IgIV peuvent entraîner une élévation de la vitesse de sédimentation érythrocytaire qui ne reflète pas une inflammation persistante[1].
Les effets secondaires des IgIV incluent les réactions à la perfusion, la fièvre, les céphalées, la méningite aseptique et une anémie hémolytique auto-immune[11]. L’anémie hémolytique peut découler des anticorps du groupe sanguin contenus dans les IgIV qui, en cas d’intensification du traitement, peut déterminer si une deuxième dose d’IgIV ou un autre médicament devra être administré[11].
Les antiplaquettaires et l’anticoagulation
Les patients qui ne sont pas à risque de saignement (numération plaquettaire supérieure à 80 x 109/L) devraient recevoir de l’acide acétylsalicylique (AAS) pendant la période de fièvre pour éviter la formation d’une thrombose artérielle[1][2]. Selon les données probantes, deux approches sont tout aussi acceptables : commencer à administrer une dose anti-inflammatoire modérée d’AAS (de 30 mg/kg/jour à 50 mg/kg/jour répartie toutes les six heures), suivie d’une dose plus faible (de 3 mg/kg à 5 mg/kg une fois par jour) une fois le patient afébrile[12][13], ou administrer une faible dose d’AAS dès le départ[14].
Si l’AAS est contre-indiqué, par exemple chez les patients atteints de l’influenza ou de la varicelle, il peut être remplacé par du dipyridamole ou du clopidogrel[1]. Il faut éviter d’utiliser des anti-inflammatoires non stéroïdiens conjointement avec l’AAS, parce qu’ils peuvent compromettre l’effet antiplaquettaire (ou anticoagulant) de l’AAS[1].
Selon le degré d’atteinte coronaire, il peut être indiqué d’administrer un double traitement antiplaquettaire ou anticoagulant[15]. L’anticoagulation s’impose en présence de gros anévrismes ou d’anévrismes géants[1]. Il est alors recommandé de consulter en surspécialité.
Les corticostéroïdes
Les données probantes relatives au rôle des corticostéroïdes pour améliorer les résultats cardiaques sont conflictuelles[2][15][16]. Après avoir consulté en rhumatologie, de fortes doses de corticostéroïdes (2 mg/kg/jour de prednisone, jusqu’à concurrence de 60 mg) sont parfois prescrites pour intensifier immédiatement le traitement ou comme traitement de deuxième intention en cas de résistance aux IgIV.
Des doses pulsées de 30 mg/kg de méthylprednisolone IV (jusqu’à concurrence de 1 000 mg/dose) sont généralement réservées au traitement du syndrome de choc lié à la MK ou du syndrome d’activation des macrophages.
Les scénarios cliniques à haut risque en cas de maladie de Kawasaki
L’algorithme de la figure 1 expose les scénarios suivants.
Figure 1. Algorithme pour l’évaluation et la prise en charge de la maladie de Kawasaki (examens facultatifs entre parenthèses)
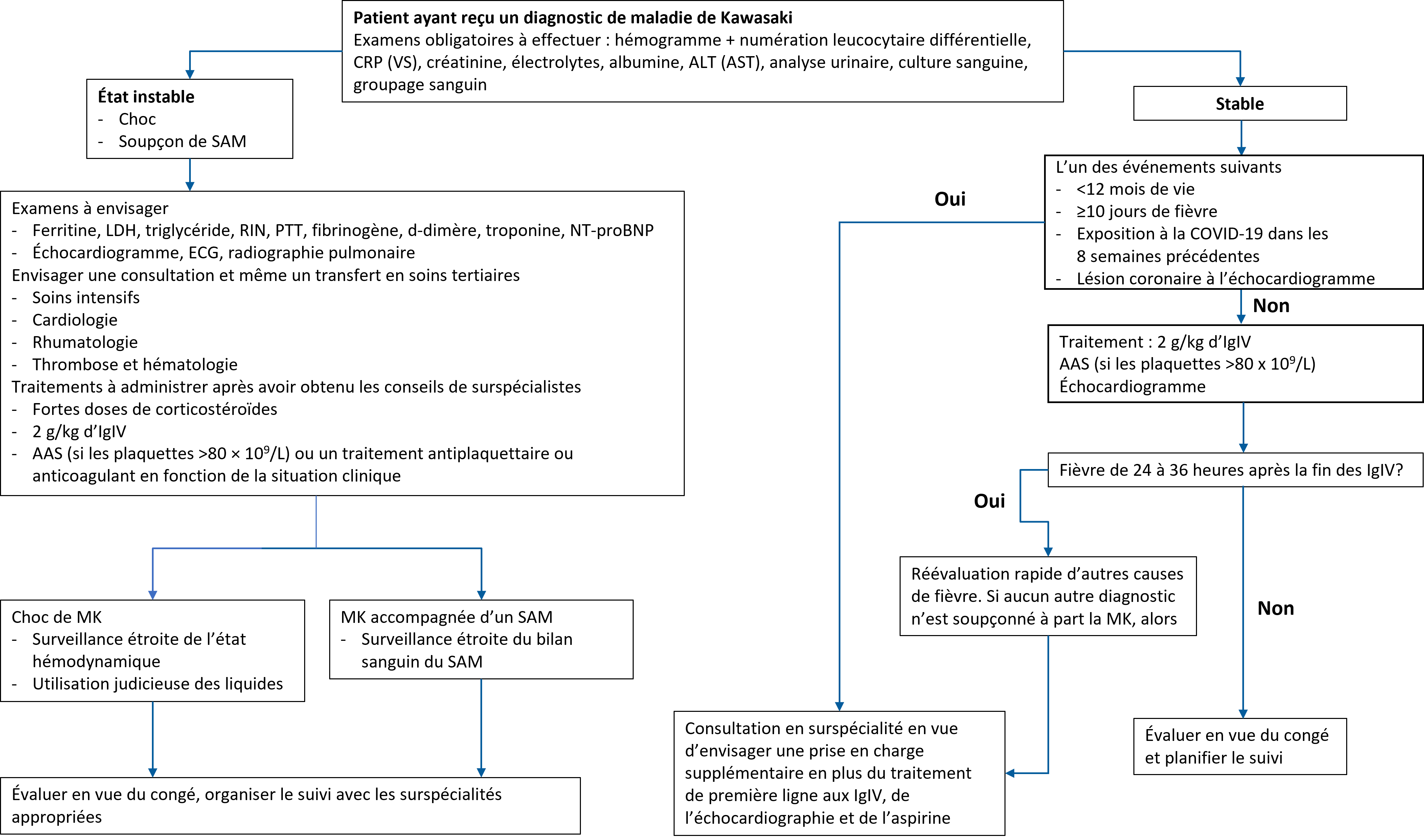
AAS acide acétylsalicylique; ALT alanine transaminase; AST aspartate transaminase; COVID-19 maladie à coronavirus 2019; CRP protéine C réactive; ECG électrocardiogramme; IgIV immunoglobulines intraveineuses; LDH lactate déshydrogénase; MK maladie de Kawasaki; NT-proBNP peptide natriurétique prohormone b n-terminale; PTT temps de thromboplastine partielle; RIN rapport international normalisé; SAM syndrome d’activation des macrophages; VS vitesse de sédimentation érythrocytaire
Le syndrome d’activation des macrophages
Le syndrome d’activation des macrophages est une affection hyperinflammatoire au potentiel fatal qui se manifeste dans 2 % à 5 % des cas de MK[17]. On doit l’envisager lorsque l’enfant est dans un état instable, qu’il ne répond pas aux IgIV ou qu’il présente une cytopénie. Une hépatosplénomégalie, une encéphalopathie et une insuffisance multiorganique en sont d’autres signes. Le bilan sanguin peut révéler une hyperferritinémie, une hypofibrinogénémie, une transaminite, une coagulopathie et une chute de la vitesse de sédimentation érythrocytaire conjointe à la hausse de la protéine C réactive[17]. La prise rapide d’un traitement immunomodulateur (p. ex., corticostéroïdes, anakinra) peut réduire considérablement la morbidité et la mortalité.
Le syndrome de choc lié à la maladie de Kawasaki
Le syndrome de choc lié à la MK désigne la totalité ou une partie des symptômes suivants : une diminution d’au moins 20 % de la tension artérielle systolique (par rapport à la normale en fonction de l’âge et du sexe) ou des signes cliniques de mauvaise perfusion périphérique[2][18]. C’est une complication rare (dans 2,6 % à 6,9 % des cas de MK), qui est probablement sous-détectée et souvent mal diagnostiquée[19]. Il est important de la détecter rapidement et de la suivre de près, car l’hypotension ou la tachycardie persistante (ou ces deux symptômes) peuvent en signaler la présence[20]. Une hydratation judicieuse est recommandée, et un soutien inotrope ou une assistance respiratoire peuvent s’imposer. Une forte dose d’attaque de méthylprednisolone peut réussir à temporiser le syndrome de choc lié à la MK[21].
Les lésions coronaires révélées à l’échocardiogramme diagnostique
Les lésions coronaires au diagnostic sont associées à un risque accru de maladie réfractaire aux IgIV qui, à son tour, serait responsable d’une augmentation de la dimension des artères coronaires[22]. Une immunosuppression supplémentaire pourrait être adoptée pour traiter une inflammation persistante. Un échocardiogramme normal à la consultation n’exclut pas la MK ni le risque de lésions coronaires après le traitement[1][2].
La résistance aux immunoglobulines intraveineuses
De 10 % à 20 % des patients atteints d’une MK résistent aux IgIV (fièvre persistante ou récurrente de 24 à 36 heures après la fin des IgIV)[23][24]. Ces patients sont plus vulnérables à un anévrisme coronaire et ont besoin d’un traitement supplémentaire[23]. Il faut également envisager d’autres causes de fièvre. Le traitement de la résistance aux IgIV est variable[25]. Une deuxième dose d’IgIV est le traitement de deuxième intention le plus fréquent[25]. Si une baisse importante de l’hémoglobine (plus de 20 g/L) est observée après la première dose d’IgIV, il faut alors envisager un autre traitement de deuxième intention (corticostéroïdes, infliximab, anakinra)[2][26][27].
D’autres caractéristiques à haut risque
Une surveillance étroite ou une intensification du traitement (ou les deux) sont à envisager en fonction de chaque cas chez les nourrissons de moins de 12 mois parce que, tout comme les enfants atteints d’une fièvre prolongée (de dix jours ou plus), ils sont les plus à risque d’anévrisme[28][29].
La maladie à coronavirus 2019
Le syndrome inflammatoire multisystémique de l’enfant est un syndrome hyperinflammatoire qu’on peut soupçonner après l’exposition à la COVID-19 ou l’infection par cette maladie au cours des deux à huit semaines précédentes et une fièvre accompagnée des caractéristiques de la MK, de symptômes gastro-intestinaux, neurologiques ou cardiaques ou d’un choc[30]-[32]. Les taux de ce syndrome ont diminué considérablement depuis le début de la pandémie[33]. La vaccination contre la maladie à coronavirus 2019 (COVID-19) demeure importante pour assurer une protection contre ce syndrome[33]. Pour en savoir plus, il suffit de consulter ce document de la SCP sur le syndrome inflammatoire multisystémique de l’enfant[32].
Le congé et le suivi
Les patients doivent être afébriles pendant au moins 24 à 48 heures après la fin du traitement aux IgIV, se soumettre à une échocardiographie de suivi et avoir accès à des soins primaires ou des soins pédiatriques pour obtenir des conseils en cas de symptômes récurrents (tableau 3). La surveillance de l’hémogramme et de la protéine C réactive de une à deux semaines après le congé peut contribuer à détecter une inflammation persistante ou une anémie hémolytique.
En général, les patients dont les artères coronaires sont normales n’ont pas besoin d’un suivi de plus de six à huit semaines[1]. Lorsqu’ils sont présents, les anévrismes petits à modérés ont habituellement régressé au bout d’un an[22]. De 1,5 % à 3 % des patients qui présentent une récidive de la MK au cours de la première année (fièvre et critères cliniques à compter de deux mois après le premier épisode)[1].
|
Tableau 3. Les directives après le congé |
|
AAS acide acétylsalicylique; IgIV immunoglobulines intraveineuses
Recommandations
- Envisager un diagnostic de maladie de Kawasaki chez les patients qui sont fiévreux et présentent moins des quatre caractéristiques principales, particulièrement s’il s’agit de nourrissons.
- Consulter en surspécialité lorsque les patients sont à haut risque de lésions coronaires ou de complications de la maladie de Kawasaki.
- En première ligne, administrer une dose de 2 g/kg d’immunoglobulines intraveineuses suivie d’un antiplaquettaire au quotidien jusqu’à la fin des échocardiogrammes de suivi.
Remerciements
Le comité de direction de la section de la pédiatrie hospitalière, de même que le comité de la pédiatrie communautaire et le comité des soins aigus de la Société canadienne de pédiatrie ont révisé le présent point de pratique.
COMITÉ DE LA PÉDIATRIE COMMUNAUTAIRE DE LA SOCIÉTÉ CANADIENNE DE PÉDIATRIE (2024-2025)
Membres : Peter Wong MD (président), Jill Borland Starkes MD (représentante du conseil), Michael Hill MD, Audrey Lafontaine MD, Meta van den Heuvel MD, Kelcie Lahey MD M. Sc.
Représentante : Richa Agnihotri MD (section de la pédiatrie communautaire de la SCP)
Auteurs principaux : Audrea Chen MD, Evelyn Rozenblyum MD, Nagib Dahdah MD, Bianca Lang MD, Rosie Scuccimarri MD, Piya Lahiry M. Sc. Ph. D. MD, Herman Tam MD, Mercedes Chan MD, Andrea Human MD, Jennifer Lee MD M. Sc., Peter Wong MD, Paul Tsoukas MD, Rae SM Yeung MD
Financement
Aucun financement n’a été accordé pour la préparation du présent manuscrit.
Conflits d’intérêts potentiels
Le docteur Rae Yeung déclare avoir reçu des fonds des Instituts de recherche en santé du Canada, de Génome Canada, de la Fondation canadienne pour l’innovation, de l’Agence de la santé publique du Canada, du Groupe de travail sur l’immunité face à la COVID-19, de Maladies rares de l’Union européenne, de Cure JM Foundation et de Maladie de Kawasaki Canada (financement révisé par les pairs) et être membre du comité consultatif médical de Maladie de Kawasaki Canada. La docteure Rosie Scuccimarri était coprésidente du comité des thérapeutiques de la Société canadienne de rhumatologie, dont elle a reçu des honoraires. Elle a été membre du comité consultatif de Novartis et a reçu un financement de Bristol Myers Squibb pour créer un registre pharmaceutique des patients. La docteure Mercedes Chan déclaré avoir reçu une rémunération de Novartis pour ses services de conseillère. Le docteur Herman Tam déclare également avoir reçu des honoraires de la Société canadienne de rhumatologie pour la préparation de contenu et de modules de formation.
Les auteurs n’ont pas d’autres conflits d’intérêts à déclarer.
Références
- McCrindle BW, Rowley AH, Newburger JW et coll. Diagnosis, treatment, and long-term management of Kawasaki disease: A scientific statement for health professionals from the American Heart Association. Circulation 2017;135(17):e927–99. doi : 10.1161/CIR.0000000000000484
- Jone P, Tremoulet A, Choueiter N et coll. Update on diagnosis and management of Kawasaki disease: A scientific statement from the American Heart Association. Circulation 2024;150(23):e481-e500. doi : 10.1161/CIR.0000000000001295
- Manlhiot C, O’Shea S, Chahal N et coll. Incidence of Kawasaki disease in Canada. Can J Cardiol 2014;30(10):S344. doi : 10.1016/j.cjca.2014.07.626
- Robinson C, Chanchlani R, Gayowsky A et coll. Incidence and short-term outcomes of Kawasaki disease. Pediatr Res 2021;90(3):670–7. doi : 10.1038/s41390-021-01496-5
- Alkanhal A, Saunders J, Altammar F et coll. Unexpectedly high incidence of Kawasaki disease in a Canadian Atlantic province—an 11-year retrospective descriptive study. Pediatr Rheumatol Online J 2023;21(1):30. doi : 10.1186/s12969-023-00805-y
- Kato H, Ichinose E, Kawasaki T. Myocardial infarction in Kawasaki disease: Clinical analyses in 195 cases. J Pediatr 1986;108(6):923–7. doi : 10.1016/s0022-3476(86)80928-3
- Tsuda E, Hirata T, Matsuo O et coll. The 30-year outcome for patients after myocardial infarction due to coronary artery lesions caused by Kawasaki disease. Pediatr Cardiol 2011;32(2):176–82. doi : 10.1007/s00246-010-9838-y
- Morishita KA, Goldman RD. Kawasaki disease recognition and treatment. Can Fam Physician 2020;66(8):577–9.
- Dallaire F, Dahdah N. New equations and a critical appraisal of coronary artery Z scores in healthy children. J Am Soc Echocardiogr 2011;24(1):60-74. doi : 10.1016/j.echo.2010.10.004
- Brown LM, Duffy CE, Mitchell C, Young L. A practical guide to pediatric coronary artery imaging with echocardiography. J Am Soc Echocardiogr 2015;28(4):379–91. doi : 10.1016/j.echo.2015.01.008
- Berard R, Whittemore B, Scuccimarri R. Hemolytic anemia following intravenous immunoglobulin therapy in patients treated for Kawasaki disease: A report of 4 cases. Pediatr Rheumatol Online J 2012;10(1):10. doi : 10.1186/1546-0096-10-10
- Sakulchit T, Benseler SM, Goldman RD. Acetylsalicylic acid for children with Kawasaki disease. Can Fam Physician 2017;63(8):607–9.
- Ito Y, Matsui T, Abe K et coll. Aspirin dose and treatment outcomes in Kawasaki disease: A historical control study in Japan. Front Pediatr 2020;8:249. doi : 10.3389/fped.2020.00249
- Dallaire F, Fortier-Morissette Z, Blais S et coll. Aspirin dose and prevention of coronary abnormalities in Kawasaki disease. Pediatrics 2017;139(6):e20170098. doi : 10.1542/peds.2017-0098
- Newburger JW. Kawasaki disease: Medical therapies. Congenit Heart Dis 2017;12(5):641–3. doi : 10.1111/chd.12502
- Green J, Wardle AJ, Tulloh RM. Corticosteroids for the treatment of Kawasaki disease in children. Cochrane Database Syst Rev 2022;5(5):CD011188. doi : 10.1002/14651858.CD011188.pub3
- Latino GA, Manlhiot C, Yeung RSM et coll. Macrophage activation syndrome in the acute phase of Kawasaki disease. J Pediatr Hematol Oncol 2010;32(7):527–31. doi : 10.1097/MPH.0b013e3181dccbf4
- Kanegaye JT, Wilder MS, Molkara D et coll. Recognition of a Kawasaki disease shock syndrome. Pediatrics 2009;123(5):e783-9. doi : 10.1542/peds.2008-1871
- Li Y, Zheng Q, Zou L et coll. Kawasaki disease shock syndrome: Clinical characteristics and possible use of IL-6, IL-10 and IFN-γ as biomarkers for early recognition. Pediatr Rheumatol Online J 2019;17(1):1. doi : 10.1186/s12969-018-0303-4
- Dionne A, Dahdah N. Myocarditis and Kawasaki disease. Int J Rheum Dis 2018;21(1):45–9. doi : 10.1111/1756-185X.13219
- Campbell AJ, Burns JC. Adjunctive therapies for Kawasaki disease. J Infect 2016;72 Suppl:S1-5. doi : 10.1016/j.jinf.2016.04.015
- Liu J, Yue Q, Qin S et coll. Risk factors and coronary artery outcomes of coronary artery aneurysms differing in size and emergence time in children with Kawasaki disease. Front Cardiovasc Med 2022;9:969495. doi : 10.3389/fcvm.2022.969495
- Tremoulet AH, Best BM, Song S et coll. Resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatr 2008;153(1):117–21. doi : 10.1016/j.jpeds.2007.12.021
- Burns JC, Capparelli EV, Brown JA et coll. Intravenous gamma-globulin treatment and retreatment in Kawasaki disease. US/Canadian Kawasaki Syndrome Study Group. Pediatr Infect Dis J 1998;17(12):1144–8. doi : 10.1097/00006454-199812000-00009
- Chan H, Chi H, You H et coll. Indirect-comparison meta-analysis of treatment options for patients with refractory Kawasaki disease. BMC Pediatr 2019;19(1):158. doi : 10.1186/s12887-019-1504-9
- Gorelik M, Lee Y, Abe M et coll. IL-1 receptor antagonist, anakinra, prevents myocardial dysfunction in a mouse model of Kawasaki disease vasculitis and myocarditis. Clin Exp Immunol 2019;198(1):101–10. doi : 10.1111/cei.13314
- Burns JC, Roberts SC, Tremoulet AH et coll. Infliximab versus second intravenous immunoglobulin for treatment of resistant Kawasaki disease in the USA (KIDCARE): A randomised, multicentre comparative effectiveness trial. Lancet Child Adolesc Health 2021;5(12):852–61. doi : 10.1016/S2352-4642(21)00270-4
- Takekoshi N, Kitano N, Takeuchi T et coll. Analysis of age, sex, lack of response to intravenous immunoglobulin, and development of coronary artery abnormalities in children with Kawasaki disease in Japan. JAMA Netw Open 2022;5(6):e2216642. doi : 10.1001/jamanetworkopen.2022.16642
- Ram Krishna M, Sundaram B, Dhanalakshmi K. Predictors of coronary artery aneurysms in Kawasaki disease. Clin Pediatr (Phila) 2014;53(6):561–5. doi : 10.1177/0009922814530802
- Tam H, Tal TE, Go E, Yeung RSM. Pediatric inflammatory multisystem syndrome temporally associated with COVID-19: A spectrum of diseases with many names. CMAJ 2020;192(38):E1093–6. doi : 10.1503/cmaj.201600
- Henderson LA, Canna SW, Friedman KG et coll. American College of Rheumatology clinical guidance for multisystem inflammatory syndrome in children associated with SARS-CoV-2 and hyperinflammation in pediatric COVID-19: Version 3. Arthritis Rheumatol 2022;74(4):e1–20. doi : 10.1002/art.42062
- Berard RA, Tam H, Scuccimarri R et coll; comité des soins aigus, Société canadienne de pédiatrie. Le syndrome inflammatoire multisystémique de l’enfant ayant un lien temporel avec la COVID-19 (mise à jour du printemps 2021). Mis à jour le 3 mai 2021.
- Yousaf AR, Lindsey KN, Wu MJ et coll. Notes from the field: Surveillance for multisystem inflammatory syndrome in children—United States, 2023. MMWR Morb Mortal Wkly Rep 2024;73(10):225-8. doi : 10.15585/mmwr.mm7310a2
Avertissement : Les recommandations du présent document de principes ne constituent pas une démarche ou un mode de traitement exclusif. Des variations tenant compte de la situation du patient peuvent se révéler pertinentes. Les adresses Internet sont à jour au moment de la publication.